婴幼儿头号遗传病杀手!一定要早筛查早预防早治疗
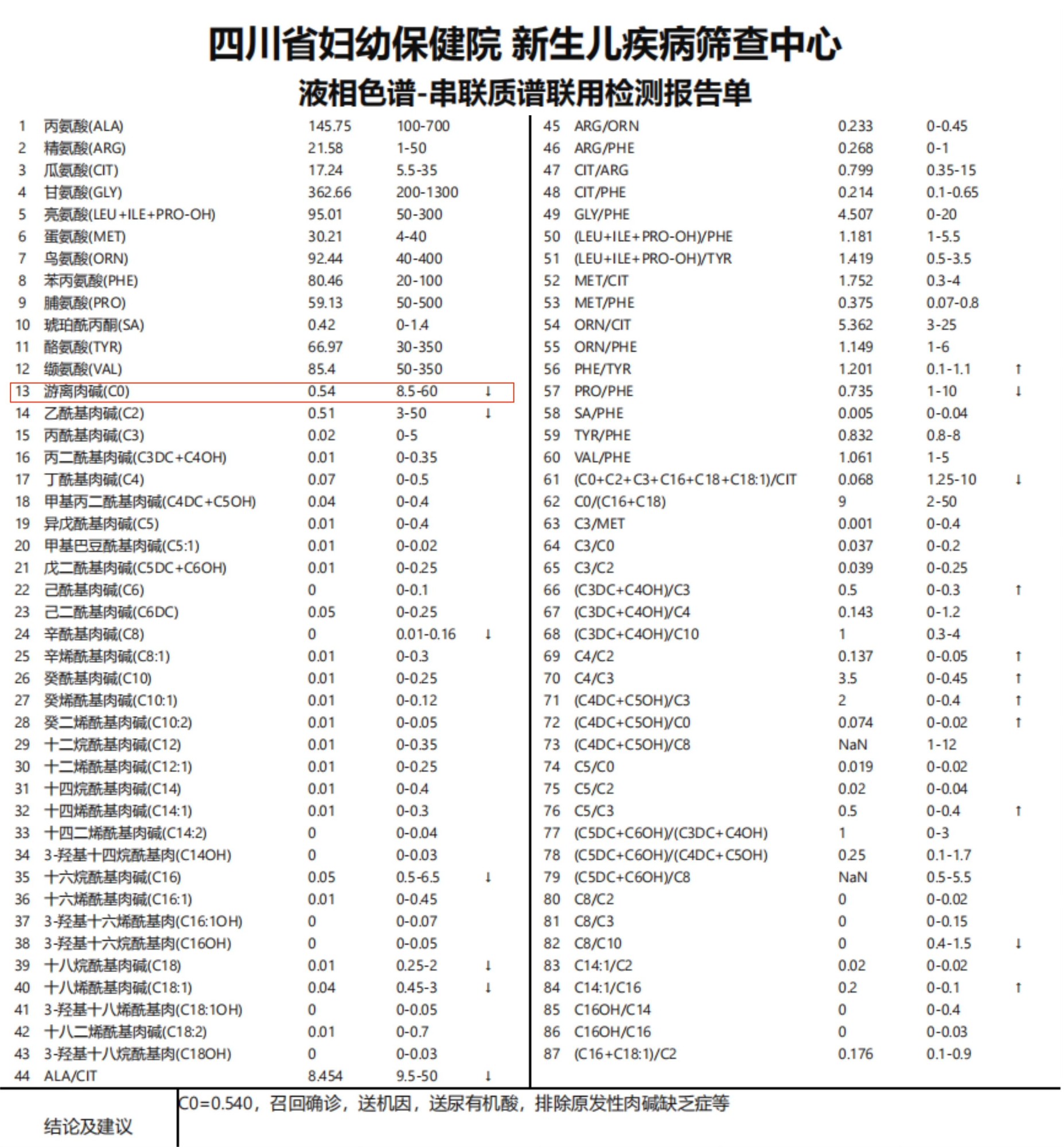
前期菠菜广告投放平台小儿神经科收治一例某地级市妇幼保健院转诊来院主诉为“肌无力”的刚满2月龄的女孩。女孩出生时可吮吸手指,但1月大时较少吮吸手指,偶尔吃奶时无力,不能抬头,几乎无蹬腿动作及上肢运动,家长没有足够重视。女孩出生时曾采足跟血筛查提示“先天性肾上腺皮质增生症”可能,做相关基因病检测提示无异常。当女孩刚满2月时,家长带小孩到当地保健院作儿保并接种“卡介苗”,医生检查发现女孩“肌张力减弱”,转到菠菜广告投放平台就诊,神经科医生询问病史了解到女孩母亲曾有一次自然流产史,查体发现女孩四肢肌力及肌张力减退,下肢重于上肢,高度怀疑女孩患上了罕见的“脊髓性肌萎缩症(SMA)Ⅰ型”。 于是,立即联系产前诊断实验室做脊髓性肌萎缩症基因检测。2天后qPCR基因检测结果显示“SMN1基因第7、8外显子纯合缺失”,女孩家长仅花费了350元检测费用就得到了确诊。由于女孩来院时合并肺炎,呼吸肌无力,经重症医学科和神经科医生紧密合作,女孩病情控制,转危为安,出院回家口服基因治疗 药物“利司扑兰口服液”,1月后(即2天前)门诊复查运动能力明显进步,继续康复治疗中。
1.什么是脊髓性肌萎缩症(SMA)?
脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA),是由于脊髓运动神经元退化死亡,导致肌无力和肌萎缩的神经肌肉遗传性疾病,发病率约1/10 000~1/6000。SMA的临床表现差异较大,依据起病年龄和病情进展不同,由重到轻分为0~Ⅳ型,其中占比最高(约45%)的SMAⅠ型患儿往往出生后6个月内发病,病情进展迅速,多因呼吸肌功能丧失导致呼吸衰竭,在两岁以内死亡,因此SMA也被称作“婴幼儿头号遗传病杀手”。国内文献报道新生儿期发病16例SMA患儿,死亡10例,均死于肺部感染并呼吸衰竭[1,2]。
2.SMA的病因是什么?
SMA是由于人体内被称为“运动神经元存活1”基因(SMN1)的缺失或异常(突变)导致的。对于正常人而言,SMN1编码产生“运动神经元存活蛋白(SMN)”,该蛋白是一种广泛表达的管家蛋白,参与多种重要的生命活动。如SMN1基因异常,人体内SMN蛋白会缺失或显著减少,进而导致运动神经元出现严重问题。根据患者发病时间及所能达到的最大运动功能指标,SMA被分为0、Ⅰ、Ⅱ、 Ⅲ、Ⅳ 5个类型(如下表),但每个SMA患者病程可能会有所差异。一般SMA不被视为一种进行性疾病,但随着肌肉不断弱化,患者表现为逐渐丧失各种身体功能。这一过程可能是渐进性的,也可能在生长突增或疾病期间明显加重,通常随着年龄增长,身体功能逐渐丧失。也有部分患者在相当长时间内,甚至数年之久,身体功能 可保持稳定[3]。

3.SMA的遗传方式是怎样的?
SMA是一种常染色体隐性遗传疾病。患儿父母通常各自有一个SMN1基因缺陷,没有患病症状,被称为携带者。如果父母双方都是缺陷基因的携带者,并各自将这一缺陷基因遗传给孩子,孩子获得了两个有缺陷的基因,就会患病,出现临床症状。最常见的SMA由5号染色体上的SMN1(运动神经元存活基因1)突变引起,因此也被称作5qSMA。通常,人群中约每50人中就有1个是SMA致病基因携带者,不同种族、地域有所不同。如父母同为该基因携带者,他们每次怀孕都有:孩子25%的几率是SMA患者;50%的几率是SMA致病基因携带者;25%的几率是健康的[3]。

4.怎样诊断SMA?
目前强调SMA 患者早诊断、早治疗,提高生存质量。从上面列表中各亚型的临床表现可以看出,Ⅰ型SMA患儿症状重、预后差。通常Ⅰ型 SMA 患儿在出生后三个月内出现快速神经元蛋白丢失,并在两岁前持续降低。尽管有机械通气等治疗方法支持生命,但是,如果没有基因药物维持治疗,Ⅰ型 SMA患儿两岁时生存率仅达到30%,其治疗时间窗非常狭窄。Ⅱ、Ⅲ 型 SMA患儿自然病程中,5岁左右可达到最大运动能力,之后运动功能逐渐下降,随着患者年龄增长,可能还会出现一系列的并发症如脊柱侧弯、关节脱位等。因此,早期发现和诊断对于各型 SMA 患儿预后至关重要,如能在出现临床症状前及时治疗,可避免机体各器官受到不可逆损害(尤其是Ⅰ型 SMA 患儿)。
SMA 临床诊疗流程,主要由以下几个环节组成:(1)临床评估:根据病史查体拟诊,主要特点为进行性、对称性四肢和躯干的肌无力,近端重于远端,下肢重于上肢;(2)辅助检查:血清肌酸激酶(CK)值正常或轻度升高,肌电图提示神经源性损害,肌活检可见大部分肌纤维呈束性萎缩;(3)分子诊断[如MLPA(多重连接探针扩增技术)、qPCR(前述病例使用)等]显示:SMN1外显子第7或第7、8纯合缺失可确诊SMA;(4)基因诊断:在SMN1外显子第7和8纯合缺失的患者中进行SMN1、SMN2拷贝数定量分析,在SMN1杂合缺失的患者中,进一步做基因序列测定;(5)基因检测阴性的患儿需行肌电图及肌肉活检进一步明确 [3]。
5.目前SMA能治疗吗?
由于SMA涉及多系统损害和并发症,所以多学科综合管理对SMA患者非常重要。菠菜广告投放平台小儿神经科设有专门的SMA治疗团队,可以对SMA患儿进行治疗和综合管理。目前国内SMA患者有两种基因治疗药物可以选择:一种是已纳入国家医保,2021年底国家医保局“灵魂砍价”引发广泛关注的诺西那生钠注射液(Nusinersen),该药物需要(腰穿)鞘内注射用药,第一年需用药6针,后续每年用药3针;另一种是口服药利司扑兰口服液(Risdiplam Powder for Oral Solution),已在我国药监局获批临床应用1年余(暂未进入医保)。这两种药物治疗均需要长期用药。根据小儿神经科多例SMA患儿(包括1家庭2例 SMA Ⅱ型患儿)的治疗经验和运动评估,两种药物均能明显提高患儿的运动能力和生活质量。
此外,2019年5月,美国FDA批准了索伐瑞韦(Zolgensma)用于临床。作为全球首个能通过一次静脉注射给药治疗SMA的药物,可用于2岁以下的患儿,并已在美国得到广泛应用。该药物每剂(一次性治疗)标价为100万美元以上(国内尚未获批)。
6.怎样预防SMA?
夫妻双方明确为SMA致病基因携带者或曾生育过SMA患儿的家庭,即为SMA高风险家庭。对于尚在备孕的高风险夫妻,可以借助胚胎植入前遗传学诊断(即三代试管婴儿)选择正常胚胎受孕,实现孕育健康宝宝的愿望。高风险家庭应在怀孕后及时到专科就诊,并按专科医生的指导尽早进行产前诊断,采集胎儿标本(如绒毛、羊水等),对胎儿进行基因诊断,评估胎儿出生后患病的风险。对普通人群SMA携带者筛 查,对SMA高风险家庭进行胚胎植入前遗传学诊断和孕期产前诊断是预防SMA患儿出生的有效途径[3,4]。
此外,大多数发达国家和国内发达地区已广泛开展新生儿SMA基因筛查(即新生儿期采集足跟血斑进行基因筛查),可在新生儿期诊断SMA患儿。国外的研究和经验表明,及早的基因治疗可以让患儿达到正常儿童相当的发育水平,明显改善疾病预后,有效减少该病的社会和家庭负担。
菠菜广告投放平台有优秀的生殖医学、产前诊断、新生儿疾病筛查和小儿神经/遗传代谢内分泌等多学科管理团队,可以全生命周期参与遗传代谢疾病的三级预防管理,满足SMA患者及家庭(包括其他遗传代谢性疾病诊治)的全部健康需求。
参考文献:
1. 刘银芝,廖镇宇,杨志明,等. 新生儿脊髓性肌萎缩症临床特征分析[J]. 中国医师杂志,2021,23(3):416-419.
2. 梁晓冰,敖当,林少珠,等. SMN1基因7号外显子纯合缺失致新生儿期发病的脊髓性肌萎缩症一例[J]. 海南医学,2022,33(6):802-804.
3.国家卫健委罕见病协作组.罕见病诊疗指南[M].北京:人民卫生出版社,2019:702-707.
4.中华医学会医学遗传学分会遗传病临床实践指南撰写组, 潘建延, 谭虎, 等.脊髓性肌萎缩症的临床实践指南 [J] .中华医学遗传学杂志,2020,37 (03): 263-268.
菠菜广告投放平台小儿神经/遗传代谢内分泌科介绍:
开展了视频脑电图、肌电图与诱发电位、基因检测、新生儿遗传代谢病筛查等检查。主要诊治儿童惊厥、运动障碍、发育障碍等各种神经/遗传代谢内分泌疾病。对癫痫、感染和免疫性脑炎、神经肌肉病、周围神经病、脑血管病、颅内肿瘤、神经心理发育、遗传代谢内分泌相关疾病、新生儿疾病筛查等各种神经/遗传代谢性疾病 积累了丰富的诊治经验。
门诊医师简介:
罗泽民,主任医师,菠菜广告投放平台神经/遗传代谢内分泌(新生儿筛查)科主任,四川省医学会小儿神经组委员, 四川省抗癫痫协会脑电图分会常务委员,四川省康复医学会儿科分会常务委员、罕见病分会委员。擅长小儿神经、遗传代谢内分泌疾病诊治。
门诊时间周一、周日全天,周三上午;周四下午特需门诊。
欧明才,主任医师,菠菜广告投放平台新生儿疾病筛查中心办公室副主任,中国妇幼保健协会遗传代谢病和维生素代谢专业委员会副主任委员、中华预防医学会出生缺陷预防与控制专委会新生儿疾病筛查学组成员、四川省医学会儿科专委会儿童内分泌遗传代谢学组委员。擅长新生儿疾病筛查业务管理和筛查疾病患儿诊治。门诊时间周一、二、六上午,周四下午。
朱书瑶 小儿神经科主治医师,擅长儿童神经系统疾病诊治。门诊时间 周二、六全天,周四上午。
蒋 琼 小儿神经科主治医师,擅长儿童神经系统疾病诊治。门诊时间 周五全天。
咨询电话:028-65978328、65978329(住院部);028 65978191(门诊)。